
Description of the Upcoming Change in Data Analysis for USP Dissolution Performance Verification Tests
Walter W. Hauck1, Anthony J. DeStefano, William E. Brown, Erika S. Stippler, Darrell R. Abernethy, Roger L. Williams, and the Biopharmaceutics Expert Committee21 Correspondence should be addressed to: Walter W. Hauck, PhD, Senior Scientific Fellow, US Pharmacopeia, 12601 Twinbrook Parkway, Rockville, MD 20852-1790; tel. 301.816.8390
2 For a list of the members of the Biopharmaceutics Expert Committee, please see Appendix A
ABSTRACT
As part of its evaluation of the performance verification tests used periodically to affirm the integrity of the USP Performance test when General Chapter Dissolution <711> is relied upon, the Biopharmaceutics Expert Committee of the Council of Experts, working with staff, decided to change the form of the accept/reject decision from one based on the result for each tablet to one based on the mean and coefficient of variation of results from a set of tablets. This paper describes the new approach. The paper also describes an implementation period for the approach, coupled with a period during which USP will discontinue use of the Salicylic Acid tablet in a performance verification test.
INTRODUCTION
USP has embarked on a vigorous program to evaluate its performance verification tests (PVTs) in order to maximize their value in ensuring the integrity of the dissolution and other procedures (1, 2). For the dissolution procedure described in General Chapter Dissolution <711> and applied to nonsolution orally administered dosage forms, this has led to two major changes.
First, Salicylic Acid tablets will be discontinued as an available official USP Reference Standard (RS) for use in a PVT. The transition is expected at the end of CY 2009. Adequate information will be made available to users to allow a smooth transition, and USP's remaining PVT tablets (Prednisone and Clorpheniramine Maleate Extended-Release RS tablets) will continue to be supplied as before.
Second, the form of the accept/reject decision for the PVT will change. A 2007 Stimuli article proposed the change from one based on per-tablet results—which correspond to individual positions in an assembly—to one based on the mean and coefficient of variation (CV) of a set of RS tablet results (3). Note that an assembly is the complete dissolution test equipment including 6 to 12 apparatus positions, depending on the manufacturer. Responses to the authors of the five comments received regarding the Stimuli article were published in a subsequent Stimuli article (4). One comment suggested that the test be done in a two-stage fashion. This is similar to the current procedure for General Chapter Dissolution <711> (5) and for General Chapter Uniformity of Dosage Units <905> (6). A two-stage test is an optional part of the proposal described here.
USP's Biopharmaceutics Expert Committee (BPC) reviewed the current proposal and concluded that USP should proceed with implementation of the revised form of the PVT acceptance criteria, as described in the 2007 Stimuli article (3), and include the option of a two-stage test. The purpose of this Stimuli article is to describe the revised approach that will apply to PVTs used to assess the integrity of the dissolution procedure as described in General Chapter <711>. The background information provided to the BPC, including operating characteristic curves, will be submitted for publication elsewhere (article in preparation).
DISCUSSION
Single-Stage Test
The current acceptance criteria for the dissolution PVT are per RS tablet. That is, the result for each tablet/position must fall within the acceptance range, which arises from collaborative studies of RS tablets and is based on both inter- and intralaboratory variability. The proposal is to replace the current approach with one based on the mean and CV of results from a set of tablets judged relative to acceptance ranges obtained from the collaborative study. The new approach will follow ISO International Standard 5725�6 (7). In Technical Specification 21748 ISO recommended a minimum of 15 degrees of freedom for the variability (8). USP elected to increase the number of tablets in the PVT from that currently required but to a lesser extent than that called for by the 15-degree-of-freedom recommendation.
The following are step-by-step instructions for the single-stage test. Sufficient detail is provided so readers can both understand the procedure and, if desired, perform the calculations. USP will make available on its Web site a spreadsheet that will accept data from the 12 to 16 individual results from steps 1 and 2 and perform all the calculations.
- For each position in the assembly, test one USP PVT RS (Prednisone RS tablets for Apparatus 1 and 2, and Chlorpheniramine Maleate Extended-Release RS tablets for Apparatus 3 at each dip rate), and record the percent dissolved at each sampling time point(s) specified for that apparatus (i.e., 30 min for Apparatus 1 and 2 and each of the times specified for Apparatus 3). After transforming the percent dissolved results to the log scale, determine the mean and variance. For assemblies with 12 positions (12 dissolution vessels), no further testing is required.
- For assemblies with fewer than 12 positions, repeat Step 1 with an additional set of tablets. Again after transforming the percent dissolved results to the log scale, determine the mean and variance.
- Calculate the average of the two means and of the two variances obtained in Steps 1 and 2. (Use the results from Step 1 alone for assemblies that have 12 positions.)
- Convert the results of Step 3 to a geometric mean (GM) and percent coefficient of variation (%CV), and round both to one decimal place following USP rounding rules (9).
- Compare the results of Step 4 to the acceptance ranges on the data sheet that is shipped with the product. The GM must not fall outside the limits, and the %CV must not be greater than the limit. If both meet the criteria, the assembly has passed the PVT.
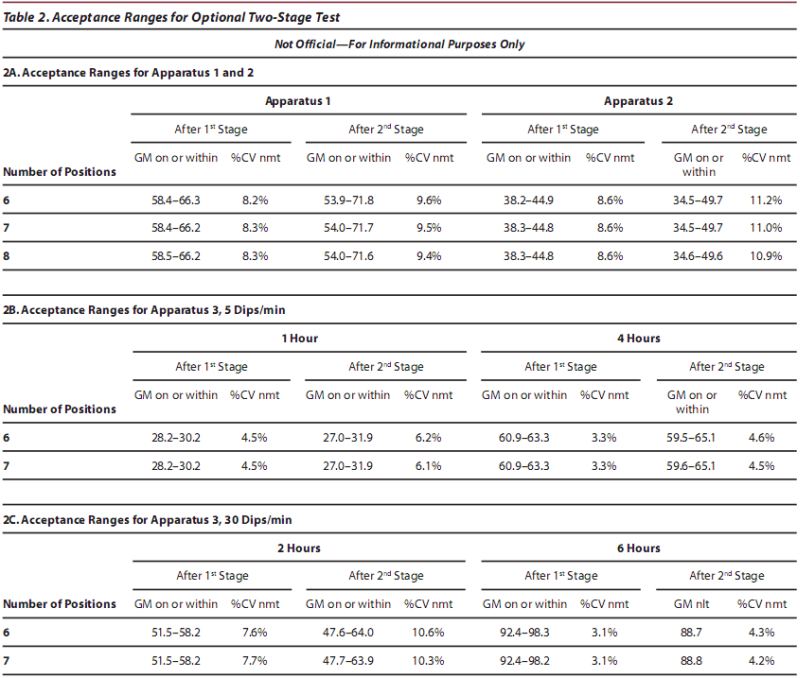
Table 1 shows what the acceptance ranges would be for the current lots of Prednisone and Chlorpheniramine Maleate Reference Standard tablets. (NOTE: The criteria for Prednisone Lot P RS tablets reflect changed ranges as of 16 July 2007.) The decision approach for Apparatus 3 uses 97.5% limits rather than the 95% limits used for the other apparatus in order to adjust for the two independent tests at the two dip rates. NOTE: These acceptance ranges are not official and are provided for information only.

Figure 1 shows a spreadsheet demonstrating the steps outlined above. The data are from an experiment using Apparatus 1. These data fail the proposed approach because the geometric mean is too low. Note that each result for the first run meets the current per-tablet criteria, but one result of the second run does not (the value of 41.1 is below the lower limit of the current range).
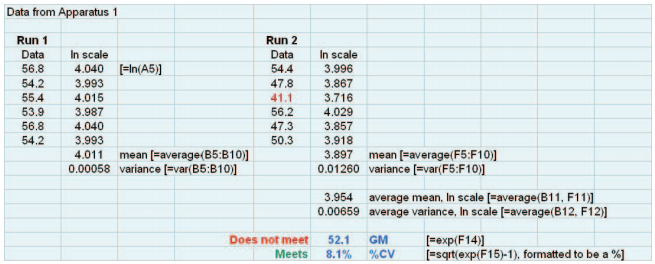
Figure 1. Spreadsheet showing implementation of the steps of the basic (single-stage) test.
For laboratories the new approach will mean less work for Apparatus 1 and 2. Because Salicylic Acid tablets are being discontinued, even if the total number of tablets remains unchanged less work remains because the assay for only one compound is required. For Apparatus 3 an increase in work is required because of the doubling in the number of tablets tested. Laboratories can reduce their work for all apparatus by choosing the optional two-stage test (see next section) and controlling their dissolution results so that the data are sufficiently tight to meet the accept/reject decision after testing only the first set of tablets.
Optional Two-Stage Test
A laboratory may choose to implement the PVT as a two-stage test. In the clinical trials literature the underlying idea that supports the two-stage test is termed a group sequential design. That is, the two-stage test is a statistically valid means of allowing the possibility of stopping the test at the first stage. A group sequential design allows for early stopping of the full 12�16 tablet test with a penalty, but the test is still considered a 12�16 tablet test. As will be seen below, the accept/reject decision rules are more stringent at the first stage (the penalty) than at the second and, at the second, slightly different from the single-stage criteria shown in Table 1. Assemblies with 12 positions are not included in the two-stage option because two stages would require users to test 24 tablets, although an assembly with 6 positions would never be required to test more than 12 tablets.
Following are step-by-step instructions for the two-stage procedure. As was the case with the single-stage test, USP will make available on its Web site a spreadsheet that practitioners can use to perform the calculations.
- For each position in the assembly, test one USP PVT RS (Prednisone RS tablets for Apparatus 1 and 2, and Chlorpheniramine Maleate Extended-Release tablets for Apparatus 3 at each dip rate), and record the percent dissolved at each sampling time point(s) specified for that apparatus (30 min for Apparatus 1 and 2 and each of the times specified for Apparatus 3). After transforming the percent dissolved results to the log scale, determine the mean and variance.
- Convert the results of Step 1 to a GM and %CV, round both to one decimal place following USP rounding rules (9), and compare to the Stage One acceptance ranges on the data sheet that is shipped with the product. The GM must not fall outside the limits, and the %CV must not be greater than the limit.
- If results of Step 2 satisfy both acceptance criteria, stop; the assembly has passed the PVT. Otherwise, continue to Step 4.
- Repeat Step 1 with an additional set of tablets and after transforming the percent dissolved results to the log scale determine the mean and variance for the data obtained at this step.
- Average the two means and two variances obtained in Steps 1 and 4.
- Convert the results of Step 5 to a GM and %CV, and round both to one decimal place following USP rounding rules (9).
- Compare the results of Step 6 to the Stage 2 acceptance ranges on the data sheet that is shipped with the product. The GM must not fall outside the limits, and the %CV must not be greater than the limit. If both meet the acceptance criteria, the assembly has passed the PVT.
Table 2 shows what the acceptance ranges would be for the two-stage test for the current lots of Prednisone and Chlorpheniramine Maleate Reference Standard tablets. NOTE: These acceptance ranges are not official and are provided for information only.
Figure 2 shows a spreadsheet implementing the procedure outlined above. The data are from Apparatus 1 and are the same as in Figure 1. These data do not meet the acceptance criteria after the first stage because of a geometric mean that falls below the Stage 1 lower limit in the new acceptance range, which requires the second stage of testing. They then fail the new acceptance criteria after the second stage because the geometric mean is below the stage 2 lower limit in the new acceptance range.
Laboratory personnel should be aware of some special considerations when they choose the two-stage test:
- First, if a second stage is required, the assembly has not failed the test after the first stage. It is still a test of 12 to 16 tablets, depending on the number of positions in the assembly. The assembly can be considered to have failed only after the second stage, e.g., only after the full complement of 12 to 16 tablets is tested.
- The laboratory has the option of stopping the test after the first stage if it sees a problem in the assembly's operation. It is not a requirement of the PVT to complete the second stage in such a circumstance. However, after taking action(s) to fix the problem, the laboratory must start the PVT again at Step 1.
- The choice of using the basic single-stage test or the two-stage test must be made as a matter of company policy and documented in standard procedures for the PVT. The choice of single- or two-stage test can differ with respect to different apparatus.
Implementation
USP is implementing two changes in its dissolution PVT. First, Salicylic Acid tablets will be discontinued as an official USP RS for use in a PVT. The discontinuation is proposed as a revision to <711> in PF 34 (5) with a targeted official date at the end of 2009. USP has sufficient inventory of the Salicylic Acid tablets to meet industry needs during this period. Adequate information will be made available to users to allow a smooth transition, and USP's remaining PVT tablets (Predisone and Clorpheniramine Maleate Extended-Release RS tablets) will continue to be supplied as before. The second is the change in the design of the PVT study, increasing from 6�12 tablets to 12�16, and the form of the accept/reject decision rule described herein. Although these are two separate decisions, USP has chosen to link their implementation in order to simplify the process of changing USP documents and laboratory SOPs. To implement both changes, USP will be publishing changes to General Chapter <711> to remove the test using Salicylic Acid and to remove the per-tablet approach. These changes are to USP text only and do not influence the harmonized portions of this chapter.
A new lot, Q, of the Prednisone RS tablets is in production. As was the case for prior lots, a collaborative study will be conducted to determine the PVT acceptance ranges. Both the new and current (per-tablet) acceptance ranges will be determined for Lot Q and will be proposed to USP's Biopharmaceutics and Reference Standards Expert Committees for endorsement and approval. The current plan is that the per-tablet accept/reject approach will continue as official at the time of release of the Lot Q series of Prednisone RS tablets in early 2009. The new accept/reject approach, as described here, will then become official for Apparatus 1 and 2 in late 2009 after a period of time during which users will have an opportunity to become more familiar with the changes. The official date for the change for Apparatus 3 has not been decided. Once the new approach becomes official for an apparatus, the per-tablet approach will no longer be valid for that apparatus. Actual implementation will be executed when USP states the new approach and acceptance ranges on the data sheets shipped with the reference standards. Removal of Salicylic Acid as a PVT test for Apparatus 1 and 2 will occur on 1 December 2009, the date when changes published in USP 32, Supplement 2, become official. USP will make available spreadsheets (compendial tools) similar to those illustrated by Figures 1 and 2. Laboratory staff will be able to enter their data in the spreadsheet, which will automatically display whether the data "meet the criteria" or "do not meet the criteria." The available USP toolkit at (10) also will be modified accordingly. USP may prepare additional training materials to assist users in the transition.
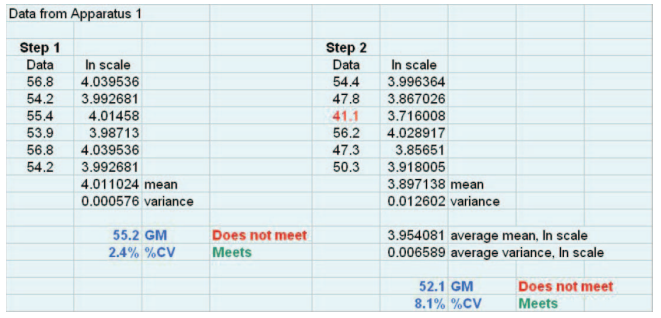
Figure 2. Spreadsheet showing implementation of the steps of the optional two-stage test.
SUMMARY
In 2009 the accept/reject approach for the dissolution PVT will change from the basis of per-tablet results to the basis of the mean and variability of results from a set of RS tablets. The change brings the USP standard into alignment with ISO International Standard 5725 (7) and advances improvement in measurement science. With the current criteria it is possible for some of the tablets to just pass at the low end of the criteria and the remainder to just pass at the high end (e.g., for Apparatus 2, three results at 30% and three at 57%). Although they technically meet the current PVT acceptance criteria, such results clearly indicate a problem with the assembly's operation/performance. With the new approach, the requirement regarding the %CV ensures sufficient consistency of results across positions in the assembly. Reliance on the mean ensures that those consistent results are within an acceptable interval of percent dissolved as determined from the RS collaborative study. The new approach will replace the current one, so individual tablet results no longer will be considered in evaluation of whether an assembly satisfies stipulations of a PVT.
REFERENCES
1. Deng, G.; Ashley, A. J.; Brown, W. E., et al. The USP performance verification test, part I: quality attributes and experimental variables contributing to dissolution variance. Pharm. Res. 2008, 25 (5), 1100�1109.
2. Glasgow, M.; Dressman, S.; Brown, W. E., et al. The USP performance verification test, part II: collaborative study of USP's Lot P Prednisone Tablets. Pharm Res. 2008, 25 (5), 1110�1115.
3. Hauck, W. W.; Manning, R. G.; Cecil, T. L.; Brown, W. E.; Williams, R. L. Proposed change to acceptance criteria for dissolution performance verification testing. Pharm. Forum 2007, 33 (3), 574�579.
4. Hauck, W. W.; Cecil, T. L.; Brown, W. E.; Abernethy, D. R.; Koch, W. F.; Williams, R. L. USP responses to comments on Stimuli article, "Proposed change to acceptance criteria for dissolution performance verification testing." Pharm. Forum 2008, 34 (2), 474�476.
5. Dissolution <711>. In United States Pharmacopeia and National Formulary USP 31�NF 26; The United States Pharmacopeial Convention, Inc.: Rockville, MD, 2008; pp 267�274.
6. Uniformity of Dosage Units <905>. In United States Pharmacopeia and National Formulary USP 31�NF 26; The United States Pharmacopeial Convention, Inc.: Rockville, MD, 2008; pp 363�369.
7. ISO. Accuracy (Trueness and Precision) of Measurement Methods and Results—Part 6: Use in Practice of Accuracy Values. ISO 5725-6:1994; Geneva, Switzerland, 1994.
8. ISO. Guidance for the use of repeatability, reproducibility, and trueness estimates in measurement uncertainty estimation. ISO/TS 21748:2004; Geneva, Switzerland, 2004.
9. General Notices, Significant Figures and Tolerances. In United States Pharmacopeia and National Formulary USP 31�NF 26; The United States Pharmacopeial Convention, Inc.: Rockville, MD, 2008; p 4.
10. Toolkit. Dissolution Procedure: Mechanical Calibration and Performance Verification Test. http://www.usp.org/USPNF/compendialTools.html (accessed Jan 13, 2009).
APPENDIX A: MEMBERS OF USP BIOPHARMACEUTICS EXPERT COMMITTEE
Thomas S. Foster, PharmD (Chair) Professor, University of Kentucky Medical Center
James E. Polli, PhD (Vice Chair) Professor, University of Maryland
Diane J. Burgess, PhD Professor of Pharmaceutics, University of Connecticut, School of Pharmacy
G. Bryan Crist, BS Scientific Affairs Manager, Varian, Inc.
Mario A. Gonzalez, PhD President, P'Kinetics International, Inc.
Vivian A. Gray, BS President, V.A. Gray Consulting, Inc.
Johannes Kraemer, PhD CEO, Phast
Lewis J. Leeson, PhD President, LJL Associates Inc.
Alan F. Parr, PharmD, PhD Director, Biopharmaceutics, GlaxoSmithKline
Leon Shargel, PhD President, Applied Biopharmaceutics
Eli Shefter, PhD Chief Scientific Officer, IriSys R & D
W. Craig Simon, PhD Manager, Bureau of Pharmaceutical Sciences Health Canada
Nhan L.Tran, PhD Senior Associate, Lachman Consultant Services, Inc.
Clarence T. Ueda, PharmD, PhD Professor of Pharmaceutical Sciences, University of Nebraska College of Pharmacy
[ Home page | About the Publication | Advertiser Links | Back Issues | Online Ordering | Calendar of Events | Current Issue | Email the editors | Editorial Advisory Board | AAPS Focus Group | USP News | Industry News | Q&A Submissions | Q&A | Resources | Article Submission Guidelines | Subscription Order Form ]